Origine des déplacements chimiques
La fréquence de résonance des protons d’une molécule va dépendre du champ B0 utilisé pour l’expérience RMN puisque : ω0 = γ.B0 . Ainsi, si un champ B0 de 9,4 T est utilisé, alors la fréquence de résonance des protons sera de l’ordre de 400 MHz.
Sur un spectre RMN il est possible de distinguer différentes fréquences de résonance. Cette différence trouve son origine dans l’environnement proche des protons qui résonnent et notamment la constante d’écran σ qui dépend des électrons proches des noyaux. Ainsi, en fonction de leur environnement, les protons ne vont pas être soumis strictement au même B0 mais à un B0 spécifique local (B0 (local)) : B0 (local) = B0 (1 – σ)
Ainsi les effets inductifs (donneur +I et attracteur -I) et les effets mésomères (donneur +M et attracteur –M) - deux effets électroniques qui s’expriment dans les composés organiques suivant les atomes présents dans la structure et leur agencement en fonction chimique - ont un impact important sur les fréquences de résonance des protons d’un composé.
Cas spécifique des protons ou groupes de protons non équivalents
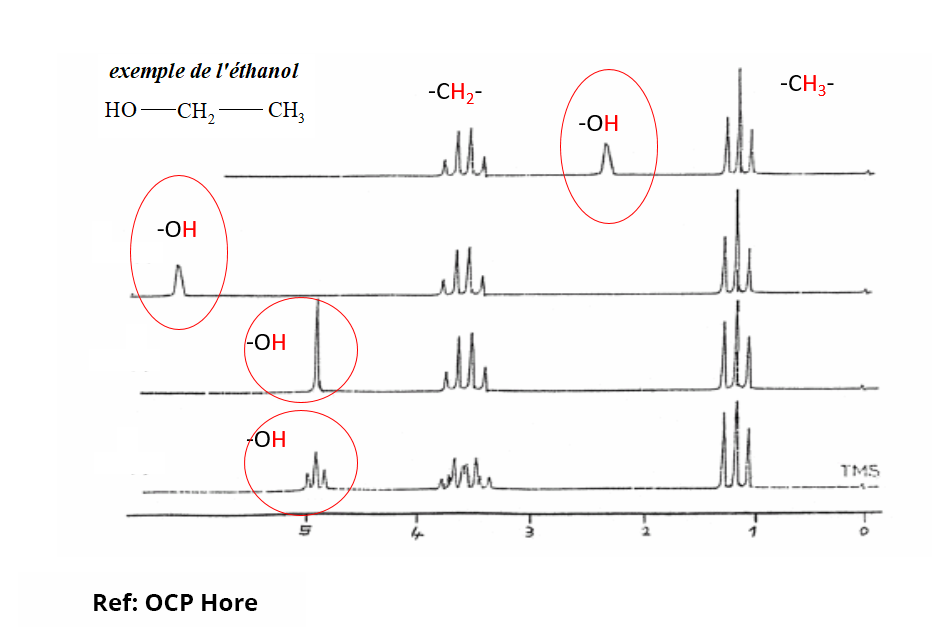
En pratique, pour exprimer la fréquence de résonance des noyaux d’intérêt en s’affranchissant de la valeur du champ B0, on utilise le déplacement chimique δ (ppm). La variation de déplacement chimique des différents protons classiquement rencontrés dans les composés organiques est de 20 ppm maximum. Il existe des compilations de déplacements chimiques expérimentaux pour différents noyaux qui aident grandement à l’analyse et à l’interprétation des spectres RMN.
Les zones de déplacement chimique sont caractéristiques des atomes d’hydrogène impliqués dans des fonctions ou environnements chimiques particuliers (zone des aliphatiques, des aromatiques, etc.). Seuls les protons labiles peuvent avoir une variation complète de leur déplacement chimique en fonction des conditions d’échantillonnage.